REPORTING AN ADVERSE EVENT OR SIDE EFFECT
It can take up to 15 years to develop a new medical treatment, with rigorous investigations carried out along the way to monitor and improve its effectiveness and safety. But we don’t stop there – we continue monitoring our medical products after they become available to patients to ensure there are not any unexpected side effects.
Side effects, also called "adverse effects" or "adverse events", refer to the unintended or undesirable events you may experience after use of a medical treatment. The side effects can sometimes be minor, but they can also involve serious health issues.
If you think you have experienced a side effect or unexpected reaction from a Galderma medication, medical device or cosmetic product, speak to your healthcare professional or contact your pharmacist as soon as you can.
To enable us to continuously monitor the safety of Galderma products, please also use Galderma's adverse events reporting system by using the form in our Contact Us page, call Galderma Special Services at 1-866-735-4137 or submit an email at contactus@galdermasupport.com.
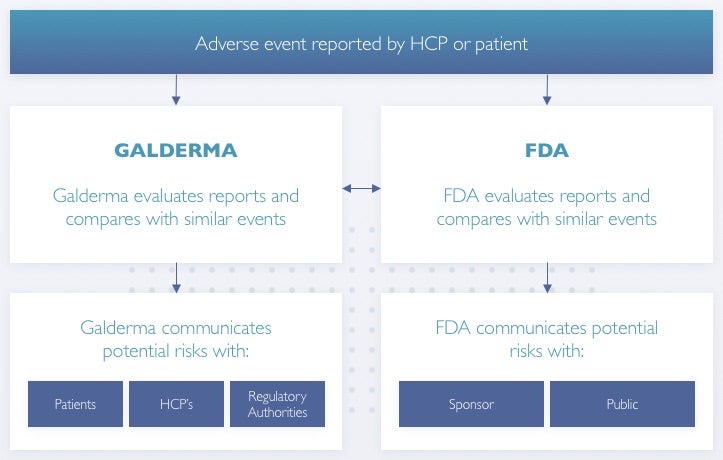
A Galderma representative will take your preliminary information. After review of this information, Galderma may follow up with you or your physician (with your permission) for additional details.
INFORMATION WE COLLECT AND WHY WE COLLECT IT
Safety information is collected in order to monitor and maintain the safety profile of all Galderma products. The laws set forth by each country's government dictate very specific requirements around the development, manufacturing and marketing of products (drugs or devices). One important part of these requirements is collection of adverse events.
PATIENTS (SUBJECT OF REPORT)
Galderma collects personal data about you when you, or a third party, provide us with information about you in relation to an adverse event. Pharmacovigilance laws require us to take “detailed records” of every adverse event reported to us, which allow the event to be evaluated and collated with other adverse events the company may have received for that same product. The personal data that we may collect about you when you are the subject of an adverse event report is:
-
name or initials;
-
age and date of birth;
-
gender;
-
weight and height;
-
details of the product causing the side effect,
-
for drug products: the dosage you have been taking or were prescribed, the reason you have been taking or were prescribed the product and any subsequent change to your usual regimen, date of treatment, date of side effect;
-
For medical devices: the type of injection, amount injected, treatment areas, date of treatment;
-
details of other medicines or remedies you are taking or were taking at the time of the side effect, including the dosage you have been taking or were prescribed, the period of time you were taking that medicine, the reason you have been taking that medicine and any subsequent change to your regimen;
-
details of the side effect you suffered, the date of side effect, the treatment you received for that side effect, and any long‐term effects the side effect has caused to your health; and
-
other medical history considered relevant by the reporter, including documents such as lab reports, medication histories and patient histories.
This information is only processed where relevant and necessary to document your side effect properly and for the purpose of meeting the requirements of our pharmacovigilance process. These requirements exist to allow us and health authorities to diagnose, manage and prevent such adverse events from occurring in the future.
REPORTERS
We collect information about you when you provide us with information about you in relation to a side effect you have reported.
Pharmacovigilance laws require us to ensure side effects are traceable and available for follow‐up. As a result, we must keep sufficient information about reporters to allow us to contact you once we have received the report. The personal data that we may collect about you when you report a side effect is your:
-
name;
-
contact details (which may include your address, e‐mail address, phone number or fax number);
-
profession (this information may determine the questions you are asked about an adverse event, depending on your assumed level of medical knowledge); and
-
relationship with the subject of the report.
Where you are also the subject of a report, this information may be combined with the information you provide in relation to your side effect.
Many health care providers worry about providing diagnostic or treatment information as a possible violation of Health Insurance Portability and Accountability Act (HIPAA) regulations, and appropriately so. However, HIPAA recognizes the need for controlled disclosure of safety data. The process of pharmacovigilance – tracking and understanding reported adverse events – is an activity where patient specific information can be shared with the pharmaceutical manufacturers (with all information protected within secure systems).
-
Health care providers are covered entities under HIPAA and may disclose protected health information to a pharmaceutical company such as Galderma, for public health purposes related to the quality, safety or effectiveness of an FDA-regulated product or activity for which that pharmaceutical company has responsibility. Examples of purposes or activities for which such disclosures may be made include, but are not limited to:
-
Collecting or reporting adverse events, product defects or problems (including problems regarding use or labeling)
-
Tracking FDA regulated products;
-
Enabling product recalls, repairs, replacement or look back (which includes locating and notifying individuals who received recalled or withdrawn products or products that are the subject of look back); and
-
Conducting post-marketing surveillance. The "person" subject to the jurisdiction of the FDA does not have to be a specific individual. Rather, it can be an individual or an entity, such as a partnership, corporation or association. Covered entities may identify the party or parties responsible for an FDA-regulated product from the product label, from written material that accompanies the product (known as labeling), or from sources of labeling, such as the Physician's Desk Reference.
Galderma communicates safety findings to regulators, patients and/or health care providers - whether favorable or unfavorable to a Galderma product. Galderma attempts to provide information appropriate to each audience in an accurate, objective and balanced manner, in order for physicians and patients to make more informed decisions about Galderma products.
It is important for Galderma, regulators, health care providers and patients to work together to ensure that all participate in the reporting of any adverse event that might be attributed to a medication or medical device.

Contact us
To enable us to continuously monitor the safety of Galderma products, please also report to Galderma the side effect you are experiencing by using the form in our Contact Us page, call Galderma Special Services at +1-866-735-4137 or submit an email at contactus@galdermasupport.com.